PK Deficiency Overview
PK deficiency is a systemic and progressive red blood cell (RBC) disorder characterized by lifelong chronic hemolysis1,2
Because of the risk of serious long-term complications, a definitive diagnosis is essential for monitoring and management decisions.3
Signs and symptoms associated with lifelong chronic hemolysis1,3-8
Lab values
- Low hemoglobin
- High ferritin
- Elevated bilirubin accompanied by jaundice
- Reticulocytosis
Common symptoms
- Extreme fatigue
- Cognitive difficulties
- Headache
Diagnosing early can help prevent the risk of complications that pose long-term burden to all PK deficiency patients, regardless of their degree of anemia.4,8

Understanding the rare disease
Dr Blinder, Dr Fertrin, and Dr Sheth discuss how they talk to their patients about PK deficiency and the different ways it can present.
It’s a severe hemolytic anemia that can be difficult to diagnose at times.
Dr Blinder
Potential complications pose long-term patient burden4,5,9
-
Aplastic crisis
-
Iron overload
-
Gallstones
-
Osteopenia and osteoporosis
-
Leg ulcers
-
Pulmonary hypertension
-
Splenomegaly
A Natural History Study of 254 patients demonstrated that patients with PK deficiency are at high risk of iron overload, regardless of their transfusion history4,8
38% of patients not receiving regular transfusions experienced iron overload*†
*As defined by ferritin >1000 ng/mL or chelation.
†The average age in the Pyruvate Kinase Deficiency Natural History Study (NHS) was 19 years (range 0.1-69.9; n=254).
The Pyruvate Kinase Deficiency NHS was funded by Agios Pharmaceuticals.
A Natural History Study of 254 patients demonstrated that patients with PK deficiency are at high risk of iron overload, regardless of their transfusion history4,8
38% of patients not receiving regular transfusions experienced iron overload*†
*As defined by ferritin >1000 ng/mL or chelation.
†The average age in the Pyruvate Kinase Deficiency Natural History Study (NHS) was 19 years (range 0.1-69.9; n=254).
The Pyruvate Kinase Deficiency NHS was funded by Agios Pharmaceuticals.
Prevalence estimates range from 3.2 to 8.5 Per million; actual prevalence may be higher due to missed diagnosis1,3,10,11
Challenges associated with diagnosing PK deficiency2,3,5,6
- Wide variability in clinical presentation
- Autosomal recessive inheritance
- Limitations of diagnostic tests
- Lack of familiarity among healthcare providers
- Incomplete diagnostic algorithms
Hemolytic anemias in order of prevalence in the US12-19
| US prevalence | Condition |
|---|---|
| >1000 in 1,000,000 |
G6PD deficiency |
| Hereditary spherocytosis | |
| Sickle cell anemia | |
| 1 to 100 in 1,000,000 |
Beta thalassemia |
| Pyruvate kinase deficiency | |
| Paroxysmal nocturnal hemoglobinuria (PNH) | |
| Extremely rare | Triosephosphate isomerase deficiency |
| Hexokinase deficiency | |
| Aldolase deficiency |
PK deficiency inheritance pattern2
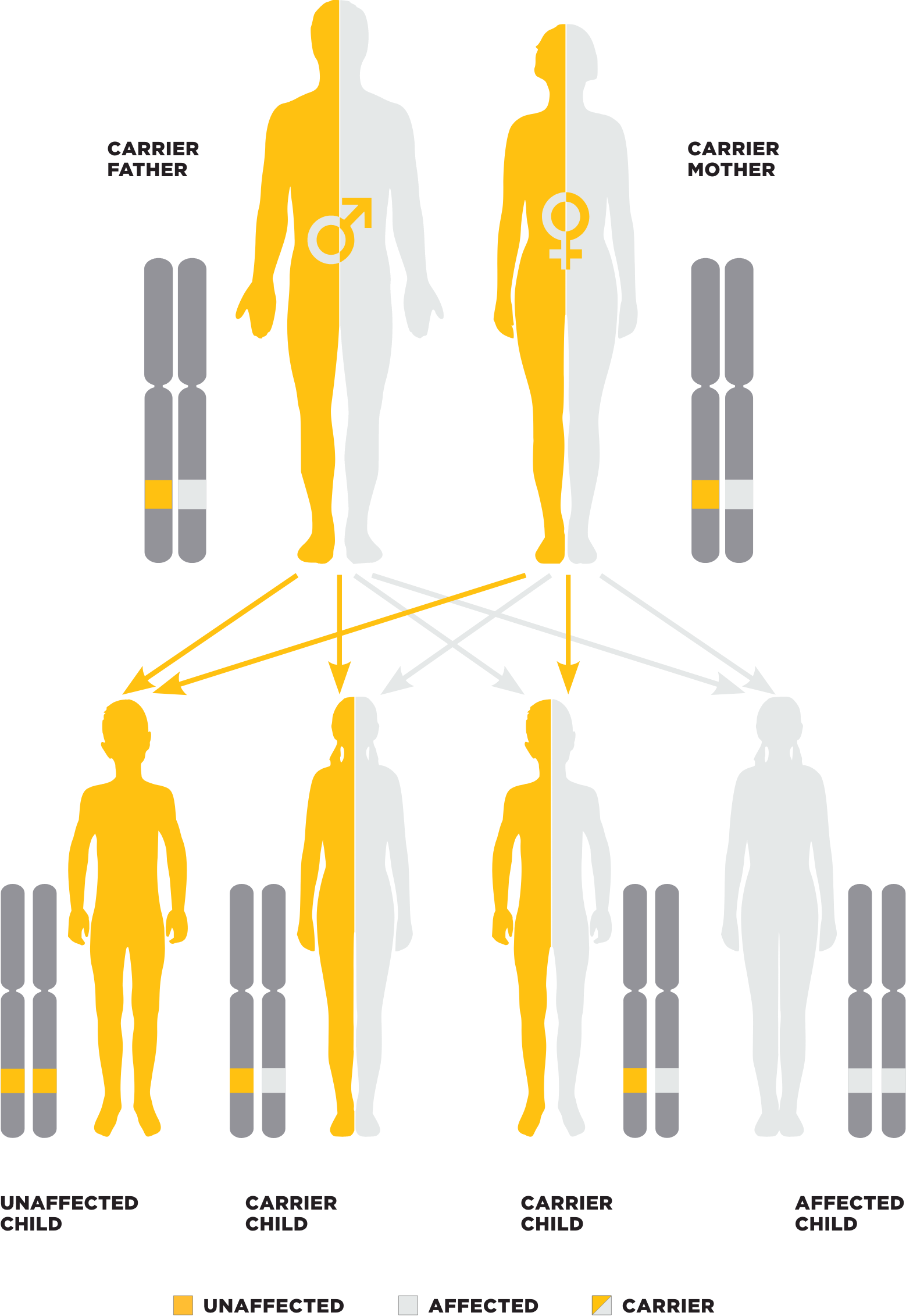
Mutations in the PKLR gene are responsible for PK deficiency1,2,4,20
- Autosomal recessive disorder; 1% to 3% of the population are believed to carry one defective copy of the PKLR gene
- Over 370 mutations identified to date
- In a recent study, approximately 35% of the pathogenic variants were previously unidentified
- The majority of patients are compound heterozygous, contributing to clinical heterogeneity
- Genotype does not always predict disease severity and patient outcomes
Patients with PK deficiency can be diagnosed at any age.
Among patients enrolled in the Pyruvate Kinase Deficiency Natural History Study, age at diagnosis ranged from 4 months to 60 years4‡
‡Age-related data available for 243 of the 254 patients.
References: 1. Grace RF, Zanella A, Neufeld EJ, et al. Erythrocyte pyruvate kinase deficiency: 2015 status report. Am J Hematol. 2015;90(9):825-830. 2. Zanella A, Fermo E, Bianchi P, Valentini G. Red cell pyruvate kinase deficiency: molecular and clinical aspects. Br J Haematol. 2005;130(1):11-25. 3. Bianchi P, Fermo E, Glader B, et al. Addressing the diagnostic gaps in pyruvate kinase deficiency: consensus recommendations on the diagnosis of pyruvate kinase deficiency. Am J Hematol. 2019;94(1):149-161 [supplementary online material]. 4. Grace RF, Bianchi P, van Beers EJ, et al. Clinical spectrum of pyruvate kinase deficiency: data from the Pyruvate Kinase Deficiency Natural History Study. Blood. 2018;131(20):2183-2192. 5. Grace RF, Cohen J, Egan S, et al. The burden of disease in pyruvate kinase deficiency: patients’ perception of the impact on health-related quality of life. Eur J Haematol. 2018;101(6):758-765. 6. Grace RF, Layton DM, Barcellini W. How we manage patients with pyruvate kinase deficiency. Br J Haematol. 2019;184(5):721-734. 7. National Organization for Rare Disorders. Voice of the Patient Report: Pyruvate Kinase Deficiency. Washington, DC: National Organization for Rare Disorders; 2020. 8. van Beers EJ, van Straaten S, Morton DH, et al. Prevalence and management of iron overload in pyruvate kinase deficiency: report from the Pyruvate Kinase Deficiency Natural History Study. Haematologica. 2019;104(2):e51-e53. 9. Boscoe AN, Yan T, Hedgeman E, et al. Comorbidities and complications in adults with pyruvate kinase deficiency. Poster presented at: The American Society of Hematology (ASH) Annual Meeting; December 7-10, 2019; Orlando, FL. 10. Carey PJ, Chandler J, Hendrick A, et al; Northern Region Haematologists Group. Prevalence of pyruvate kinase deficiency in a northern European population in the north of England. Blood. 2000;96(12):4005-4006. 11. de Medicis E, Ross P, Friedman R, et al. Hereditary nonspherocytic hemolytic anemia due to pyruvate kinase deficiency: a prevalence study in Quebec (Canada). Hum Hered. 1992;42(3):179-183. 12. Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol Dis. 2009;42(3):267-278. 13. Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372(9647):1411-1426. 14. Brousseau DC, Panepinto JA, Nimmer M, Hoffmann RG. The number of people with sickle-cell disease in the United States: national and state estimates. Am J Hematol. 2010;85(1):77-78. 15. World Health Organization, Thalassaemia International Federation. Management of haemoglobin disorders: report of a joint WHO-TIF meeting. November 16-18, 2007; Nicosia, Cyprus. 16. Centers for Disease Control and Prevention. Thalassemia awareness. https://www.cdc.gov/ncbddd/thalassemia/features/international-thalassemia.html. Reviewed April 15, 2020. Accessed May 25, 2023. 17. Secrest MH, Storm M, Carrington C, et al. Prevalence of pyruvate kinase deficiency: a systematic literature review. Eur J Haematol. 2020;105(5):173-184. 18. Dingli D, Luzzatto L, Pacheco JM. Neutral evolution in paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci U S A. 2008;105(47):18496-18500. 19. Koralkova P, van Solinge WW, van Wijk R. Rare hereditary red blood cell enzymopathies associated with hemolytic anemia—pathophysiology, clinical aspects, and laboratory diagnosis. Int J Lab Hematol. 2014;36(3):388-397. 20. Bianchi P, Fermo E, Lezon-Geyda K, et al. Genotype-phenotype correlation and molecular heterogeneity in pyruvate kinase deficiency. Am J Hematol. 2020;95(5):472-482.